Телемедицина – детям
Квалиметрия в медицине
Бессимптомная диагностика
Неинвазивное обследование
Диагностика на ранних стадиях
Обследование не выходя из дома
Стационарзамещающие технологии
Электронная диспансеризация
Узнайте о здоровье ребёнка
Комплексное обследование
Индивидуальный подход
Всё о детском энурезе
Клинические рекомендации (протоколы, руководства лечения)
"Болезнь Помпе"
(Clinical Guidelines)
Дата утверждения (частота пересмотра): 2019
По состоянию на 23.01.2023 на сайте МЗ РФ
Пересмотр не позднее: 2021
Официально применяется с 01.01.2022 в соответствии с Постановлением Правительства РФ от 17.11.2021 N 1968
Возрастная категория: Взрослые, Дети
Разработчик: Ассоциация медицинских генетиков, Союз педиатров России, РОО "Общество специалистов по нервно-мышечным заболеваниям"
Кем утверждены: Минздравом РФ
Одобрены Научно-практическим Советом Минздрава РФ
44 страницы А4
Со всеми Приложениями
Гликогеноз II типа, или болезнь Помпе (БП), относится к редким наследственным болезням накопления (OMIM # 232300), связанным с дефицитом фермента кислой мальтазы (кислой альфа-глюкозидазы, КАГ) в лизосомах. Преимущественное накопление гликогена отмечено в скелетных мышцах, но в разной степени может обнаруживаться и в других органах и тканях, включая сердечную мышцу, печень, нервную систему, гладкую мускулатуру и т.п. В литературе используются следующие общепринятые синонимы БП: болезнь накопления гликогена II типа (GSD-II); дефицит кислой мальтазы (AMD); гликогеноз II типа.
Содержание (разделы, оглавление) документа:
Разработчик
Список сокращений
Термины и определения
1. Краткая информация по заболеванию или состоянию (группы заболеваний или состояний)
1.1. Определение заболевания или состояния (группы заболеваний или состояний)
1.2. Этиология и патогенез заболевания или состояния (группы заболеваний или состояний)
1.3. Эпидемиология заболевания или состояния (группы заболеваний или состояний)
1.4. Особенности кодирования заболевания или состояния (группы заболеваний или состояний) по Международной статистической классификации болезней и проблем, связанных со здоровьем
1.5. Классификация заболевания или состояния (группы заболеваний или состояний)
1.6. Клиническая картина заболевания или состояния (группы заболеваний или состояний)
Клинические проявления младенческой формы болезни Помпе
Клинические проявления болезни Помпе с поздним началом (БППН)
2. Диагностика заболевания или состояния (группы заболеваний или состояний) медицинские показания и противопоказания к применению методов диагностики
2.1. Жалобы и анамнез
2.2. Физикальное обследование
2.3. Лабораторные диагностические исследования
2.4. Инструментальные диагностические исследования
2.5. Иные диагностические исследования
3. Лечение, включая медикаментозную и немедикаментозную терапии, диетотерапию, обезболивание, медицинские показания и противопоказания к применению методов лечения
3.1. Патогенетическое лечение
3.2. Симптоматическое лечение
3.3. Иное лечение
4. Медицинская реабилитация, медицинские показания и противопоказания к применению методов реабилитации
5. Профилактика и диспансерное наблюдение, медицинские показания и противопоказания к применению методов профилактики
6. Организация оказания медицинской помощи
7. Дополнительная информация (в том числе факторы, влияющие на исход заболевания или состояния)
Критерии оценки качества медицинской помощи
Список литературы
Приложение А1. Состав рабочей группы по разработке и пересмотру клинических рекомендаций
Приложение А2. Методология разработки клинических рекомендаций
Целевая аудитория данных клинических рекомендаций
Шкала оценки уровней достоверности доказательств (УДД) для методов диагностики (диагностических вмешательств)
Шкала оценки уровней достоверности доказательств (УДД) для методов профилактики, лечения и реабилитации (профилактических, лечебных, реабилитационных вмешательств)
Шкала оценки уровней убедительности рекомендаций (УУР) для методов профилактики, диагностики, лечения и реабилитации (профилактических, диагностических, лечебных, реабилитационных вмешательств)
Порядок обновления клинических рекомендаций
Приложение А3. Справочные материалы, включая соответствие показаний к применению и противопоказаний, способов применения и доз лекарственных препаратов, инструкции по применению лекарственного препарата
Приложение Б. Алгоритмы действий врача
Приложение В. Информация для пациента
Болезнь Помпе
Наследование
Как устанавливают диагноз
Лечение
Медико-генетическая консультация
Помощь семьи
Приложение Г1-ГN. Шкалы оценки, вопросники и другие оценочные инструменты состояния пациента, приведённые в клинических рекомендациях
Приложение Г1. Основные симптомы МБП
Приложение Г2. Основные клинические симптомы БППН
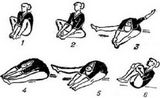
Приложение Г3. Выполнение двигательных тестов при БППН
Приложение Г4. Основные заболевания для дифференциального диагноза БППН
Приложение Г5. Мониторинг пациента с БП
Приложение Г6. Шкала Альберта моторного развития младенцев
Шкала Альберта моторного развития младенцев
Шкала оценки моторного развития у младенцев
Оценка результатов обследования по шкале Альберта моторного развития ребёнка
Приложение Г7. Сила мышц по шкале комитета медицинских исследований
Приложение Г8. 6MWT – тест 6-минутной ходьбы
Приложение Г9. Опросник качества жизни SF-36
Приложение Г10. Забор биоматериала для диагностики в пятнах крови
Алгоритм действий медицинского персонала при взятии образцов крови
Особенности при инфузионной терапии
Хранение и транспортировка биоматериала
Ферментная заместительная терапия – лечение, заключающееся во введении препарата (рекомбинантного фермента) пациентам с наследственным нарушением метаболизма. Лизосомные болезни накопления – группа наследственных моногенных заболеваний, связанных с нарушением функции лизосомю.
БП обусловлена недостаточностью фермента кислой альфа-глюкозидазы (КАГ), который относится к группе лизосомных гидролаз. Ген GAA, кодирующий КАГ, локализован на длинном плече 17 хромосомы (17q25.2-q25.3), состоит из 20 экзонов и имеет размер около 20000 пар нуклеотидов. Идентифицировано более 580 мутаций гена и их число постоянно растёт. Хотя некоторые мутации гена GAA встречаются преимущественно в определённой этнической группе, для большинства популяций отсутствуют так называемые "мажорные", составляющие сколько-нибудь значимую долю всех мутаций популяции и потому целесообразные для прицельного выявления. Мутации GAA приводят к разной степени дефицита фермента. Младенческая форма БП (МБП) развивается при значительном снижении или полном отсутствии активности КАГ, БП с поздним началом (БППН) – при менее выраженном дефиците активности КАГ, обусловленном "мягкими" мутациями гена GAA.
Отложение или утилизация гликогена зависят от потребности организма в глюкозе. Биохимические превращения гликогена в печени способствуют поддержанию нормального уровня глюкозы в крови. В скелетных мышцах при метаболизме гликогена образуется глюкозо-6-фосфат, участвующий в процессе окисления и продукции энергии, необходимой для нормальной работы мышц (ее сокращения и расслабления). Отсутствие или значимый дефицит КАГ приводит к массивному накоплению гликогена в лизосоме и нарушению функции клеток. При БП, независимо от формы, гликоген может накапливаться практически в любых тканях, но при этом имеется преимущественное скопление гликогена в разных органах и тканях, которое уже зависит от формы болезни. Так, при МБП гликоген накапливается в скелетной мускулатуре, сердечной мышце, печени, мышцах языка. Реже аномальные отложения гликогена могут встречаться в мышечном слое сосудистой стенки, определяя развитие аневризм и мальформаций, а также – в клетках центральной и периферической нервной системы. При БППН, в отличие от МБП, больше всего страдает скелетная мускулатура, в то время, как поражение остальных органов и тканей встречается значительно реже и по тяжести поражения не сопоставимо с МБП. БП характеризуется неуклонно прогрессирующим течением с разными вариантами прогрессирования. Диагностика БП, независимо от формы, основана на оценке клинических симптомов, данных инструментальных и лабораторных обследований.
Точная частота БП неизвестна. По данным разных авторов частота болезни, в зависимости от страны и этнической принадлежности, варьируют в диапазоне от 1:40000 до 1:300000. Например, в южном Китае и на Тайване частота классической младенческой (инфантильной) формы БП составляет 1:40000-50000, являясь самым частым гликогенозом. При рассмотрении данных по Тайваню отдельно, частота заболеваемости составила 1:33134. По результатам скрининга новорожденных по сухому пятну крови (СПК) в Австрии частота БП составила 1:8684. В Голландии частота гликогеноза II типа у младенцев составляет 1:138000, а формы БП с поздним началом – 1:57000.
Особенности кодирования заболевания или состояния (группы заболеваний или состояний) по Международной статистической классификации болезней и проблем, связанных со здоровьем: Согласно МКБ 10, заболевание относится к классу IV, болезням эндокринной системы, расстройству питания и нарушению обмена веществ, E74.0 – болезни накопления гликогена (болезнь Помпе).
БП относится к гликогенозам – группе редких наследственных болезней нарушения метаболизма гликогена, связанных с изменением нескольких ферментов, вовлеченных в синтез и распад гликогена. Клинические появления болезни связаны с патологическим накоплением гликогена и продуктов его метаболизма в клетках. Сегодня выделяют восемь основных типов гликогенозов.
Всех пациентов с БП, независимо от времени начала, отличает неуклонно прогрессирующий характер течения болезни. Продолжающееся отложение гликогена в тканях-мишенях нарушает их функцию и, в конечном итоге, приводит к необратимым структурным изменениям тканей и гибели больного БП. БП характеризуется полиорганной патологией, но в зависимости от времени манифеста частота вовлечения тех или иных органов и систем также будет разной. Попытки классификации БП по данным литературы в зависимости от возраста начала, с выделением младенческой формы с дебютом на первом году жизни (с подразделением на раннюю, позднюю младенческую форму, форму с/без кардиомиопатии и т.п.), детскую, ювенильную и взрослую формы болезни не нашли единодушной поддержки специалистов. С учётом единого патогенеза БП, выделяют только два варианта в зависимости от времени манифеста симптомов: Младенческая (инфантильная) БП (МБП) манифестирующая в период новорожденности или младенческом возрасте и БП с поздним началом (БППН).
Каждая из выделенных форм имеет свои особенности клиники, алгоритмов диагностики и тактики ведения пациентов. МБП характеризуется тяжелым прогрессирующим течением и быстрым развитием полиорганной патологии – мышечной гипотонии и слабости, сердечной недостаточностью в результате гипертрофической кардиомиопатии, дыхательной недостаточностью на фоне слабости диафрагмы и межреберных мышц, нарушениями питания (трудности при вскармливании) из-за слабости лицевой мускулатуры и увеличения языка, увеличением печени. Смерть при МБП чаще всего наступает на первом году жизни от сердечно-дыхательной недостаточности.
БППН отличается от МБП более мягкими клиническими проявлениями и течением, отсутствием полиорганной патологии (поражение сердца крайне редко) и более поздними осложнениями со стороны дыхательной системы в результате слабости мышц диафрагмы и межреберной мускулатуры. Обычно пациенты погибают от дыхательной недостаточности и инфекционных легочных осложнений. Время гибели пациентов при БППН зависит от момента начала и последующего характера течения болезни и может наступить в детстве, юношеском, взрослом или преклонном возрасте. Более подробно клиническая картина выделенных форм рассмотрена ниже.
Диагноз БП устанавливается на основании совокупности: анамнестических данных, клинических данных, результатов лабораторного исследования (биохимического и молекулярно-генетического анализа).
Детям иногда сложно говорить, так как страдают все группы мышц, речь становится невнятной, смазанной. Мышечная слабость может проявляться в неуклюжести, пациенты спотыкаются, им трудно подниматься по лестнице. Походка взрослых пациентов с болезнью Помпе – своеобразная, они стараются компенсировать дефект за счёт переноса центра тяжести. Чем меньше пациент двигается, тем больше вероятность развития искривлений позвоночника и появления контрактур.
Наследуется болезнь Помпе по аутосомно-рецессивному типу. Больной БП ребёнок наследует по одному измененному гену (гена с мутацией в своей последовательности) от каждого из родителей. В семьях, где родители являются носителями болезни Помпе, риск рождения больного БП ребенка составляет 25% на каждую беременность. Поэтому если в семье родился больной БП ребенок, это не означает, что все дети будут больны. Есть шанс родить здорового ребёнка.
Врачи на основании клинических симптомов могут заподозрить болезнь. Затем проводятся лабораторные тесты и инструментальное исследование. Практически у всех больных БП в биохимическом анализе крови повышена активность ферментов креатинфосфокиназы, Алат, Асат. При проведении электронейромиографии определяют первично мышечный характер нарушения. Но эти симптомы могут встречаться и при других наследственных формах мышечных дистрофий. Для точного подтверждения диагноза необходимо определение активность альфа глюкозидазы в крови. Во многих случаях также рекомендуется проведение ДНК диагностики, если активность фермента была снижена.
Для лечения БП разработана специальная ферментная заместительная терапия. Смысл терапии заключается в том, что пациенту каждые две недели внутривенно вводят недостающий фермент. Препарат для ферментной заместительной терапии называется Алглюкозидаза альфа. В самой процедуре внутривенного введения фермента нет ничего сложного и страшного. При определённом навыке это можно проводить в любой больнице. Эффект терапии во многом зависит от того, когда было начато лечение. Если процесс в мышечной ткани дошел до определённой стадии, восстановить утраченную функцию мышце невозможно.
Семьям очень важно посетить врача-генетика. У врача-генетика можно узнать риск рождения больного БП ребёнка в данной семье, пройти обследование родственникам, если это необходимо. Обсудить пренатальную и преимплантационную диагностику. Пренатальная диагностика проводится на 9-11 неделях беременности. В материале, который называется ворсины хориона (то, из чего в последующем формируется плацента), определяют активность фермента и проводят тестирование для выявления мутаций в гене GAA. На основании проведённого анализа делают вывод, болен плод или здоров. Современные технологии позволяют проводить и преимплантационную диагностику. Оплодотворение проводится в пробирке, затем отбирают только те оплодотворенные эмбрионы на стадии нескольких бластомеров, в которых нет семейной мутации, и их имплантируют в организм матери. При данной процедуре есть свои риски, которые может разъяснить врач-генетик. Болезнь Помпе не входит в перечень орфанных заболеваний, лечение которых проводится за счёт средств региональных бюджетов, однако, пациенты имеют возможность лечиться в России.
Не забывайте: от семьи зависит успех лечения не в меньшей степени, чем от врача. Нужно соблюдать рекомендации, ни при каких условиях не терять надежду. И даже если вы не победите болезнь полностью, изменить жизнь к лучшему, сделать родного вам человека счастливым в ваших силах.
Дорогие и уважаемые коллеги, посетители сайта, друзья! В 2020 году мы начали на сайте большой и важный проект – размещение новых клинических рекомендаций (КР) 2020-2021-2022-2023-2024-2025 годов выпуска. Мы стараемся делать их вёрстку максимально удобной для широкого пользования и в то же время максимально экономичной по весу в мегабайтах. Пожалуйста, поддержите нас в этой проектной инициативе любой возможной для вас суммой. Заранее благодарим и Спасибо!