|
Клинические рекомендации (протоколы, руководства лечения)
"Мукополисахаридоз тип I"
(Clinical Guidelines)
Дата утверждения (частота пересмотра): 2021
По состоянию на 19.01.2023 на сайте МЗ РФ
Пересмотр не позднее: 2023
Официально применяется с 01.01.2022 в соответствии с Постановлением Правительства РФ от 17.11.2021 N 1968
Возрастная категория: Дети
Разработчик: Союз педиатров России, Ассоциация медицинских генетиков
Кем утверждены: Минздравом РФ
Одобрены Научно-практическим Советом Минздрава РФ
51 страница А4
Со всеми Приложениями
Мукополисахаридоз I типа (МПС I) - наследственная лизосомная болезнь накопления, обусловленная дефицитом фермента альфа-L-идуронидазы и протекающая с различными клиническими проявлениями: задержкой роста, умственной отсталостью, поражением нервной системы, сердечно-лёгочными нарушениями, гепатоспленомегалией, множественными дизостозами, помутнением роговицы. Все вышеперечисленные признаки приводят к инвалидизации, а при тяжёлом течении болезни - к летальному исходу.
Содержание (разделы, оглавление) документа:
- Список сокращений
- Термины и определения
- 1. Краткая информация по заболеванию или состоянию (группы заболеваний или состояний)
- 1.1. Определение заболевания или состояния (группы заболеваний или состояний)
- 1.2. Этиология и патогенез заболевания или состояния (группы заболеваний или состояний)
- 1.3. Эпидемиология заболевания или состояния (группы заболеваний или состояний)
- 1.4. Особенности кодирования заболевания или состояния (группы заболеваний или состояний) по Международной статической классификации болезней и проблем, связанных со здоровьем
- 1.5. Классификация заболевания или состояния (группы заболеваний или состояний)
- 1.6. Клиническая картина заболевания или состояния (группы заболеваний или состояний)
- Мукополисахаридоз тип I S – лёгкая форма
- Мукополисахаридоз тип I H/S – промежуточная форма
- Мукополисахаридоз I H – тяжёлая форма
- 2. Диагностика заболевания или состояния (группы заболеваний или состояний) медицинские показания и противопоказания к применению методов диагностики
- 2.1. Жалобы и анамнез
- 2.2. Физикальное обследование
- 2.3. Лабораторные диагностические исследования
- 2.4. Инструментальные диагностические исследования
- 2.5. Иные диагностические исследования
- 3. Лечение, включая медикаментозную и немедикаментозную терапии, диетотерапию, обезболивание, медицинские показания и противопоказания к применению методов лечения
- 3.1. Патогенетическое лечение
- Трансплантация гемопоэтических стволовых клеток
- 3.2. Симптоматическое лечение
- 3.3. Хирургическое лечение
- 4. Медицинская реабилитация, медицинские показания и противопоказания к применению методов реабилитации
- 5. Профилактика и диспансерное наблюдение, медицинские показания и противопоказания к применению методов профилактики
- 5.1. Пренатальная диагностика МПС I
- 5.2. Диспансерное наблюдение
- 6. Организация оказания медицинской помощи
- 6.1. Показания для госпитализации в медицинскую организацию
- 6.2. Показания к выписке пациента из медицинской организации
- 7. Дополнительная информация (в том числе факторы, влияющие на исход заболевания или состояния)
- Критерии оценки качества медицинской помощи
- Список литературы
- Приложение А1. Состав рабочей группы по разработке и пересмотру клинических рекомендаций
- Приложение А2. Методология разработки клинических рекомендаций
- Целевая аудитория данных клинических рекомендаций
- Шкала оценки уровней достоверности доказательств (УДД) для методов диагностики (диагностических вмешательств)
- Шкала оценки уровней достоверности доказательств (УДД) для методов профилактики, лечения и реабилитации (профилактических, лечебных, реабилитационных вмешательств)
- Шкала оценки уровней убедительности рекомендаций (УУР) для методов профилактики, диагностики, лечения и реабилитации (профилактических, диагностических, лечебных, реабилитационных вмешательств)
- Экономический анализ
- Метод валидизации рекомендаций
- Описание метода валидизации рекомендаций
- Консультация и экспертная оценка
- Рабочая группа
- Порядок обновления клинических рекомендаций
- Приложение А3. Справочные материалы, включая соответствие показаний к применению и противопоказаний, способов применения и доз лекарственных препаратов, инструкции по применению лекарственного препарата
- Приложение А3.1. Нормативные документы, использованные при подготовке клинических рекомендаций
- Основные нормативно-правовые акты, регулирующие оказание паллиативной медицинской помощи
- Приложение А3.2. Классификация мукополисахаридозов
- Приложение А3.3. Выраженность клинических проявлений МПС I в разном возрасте
- Приложение А3.4. Частота проведения обследования у пациентов с МПС I типа
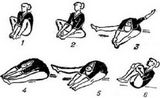
- Приложение А3.5. Тест 6 минутной ходьбы (6MTX)
- Приложение А3.6. Основные мероприятия при оказании помощи пациентам с аллергической реакцией на препарат
- Приложение А3.7. Основные мероприятия при оказании помощи пациентам с анафилактической реакцией
- Приложение А3.8. Забор биоматериала для диагностики в пятнах крови
- Алгоритм действий медицинского персонала при взятии образцов крови
- Особенности при инфузионной терапии
- Хранение и транспортировка биоматериала
- Приложение А3.9. Расшифровка примечаний
- Приложение Б. Алгоритмы действий врача
- Приложение В. Информация для пациента
- Синдром Гурлер, Шейе и Гурлер-Шейе
- Патогенез
- Наследование синдрома Гурлер, Шейе и Гурлер-Шейе
- Как устанавливают диагноз
- Клинические проявления мукополисахаридоза I типа
- Раннее развитие, рост
- Внешние особенности
- Скелет, опорно-двигательная и костно-суставная система
- Органы дыхания
- Ротовая полость и зубы
- Сердечно-сосудистая система
- Брюшная полость
- Нервная система
- Орган зрения
- Органы слуха
- Лечение, наблюдение и плановые обследования
- Наблюдение, плановые обследования и тесты
- Магнитная резонансная томография
- Рентгенография
- Симптоматическое лечение
- Скелет и опорно-двигательная система костно-суставная система
- Хирургическое лечение
- Органы слуха
- Сердечно-сосудистая система
- Инфекции
- Диетотерапия
- Патогенетическая терапия
- Трансплантация костного мозга/гемопоэтических стволовых клеток
- Половая зрелость и брак
- Отдых
- Узнать больше
- Общественные организации и фонды России
- Приложение Г1-ГN. Шкалы оценки, вопросники и другие оценочные инструменты состояния пациента, приведённые в клинических рекомендациях
Ферментная заместительная терапия - лечение, заключающееся во введении препарата (рекомбинантная человеческая альфа-L-идуронидаза) пациентам с наследственным нарушением метаболизма. Лизосомные болезни накопления - группа наследственных моногенных заболеваний, связанных с нарушением функции лизосом. Мукополисахаридозы (МПС) - группа наследственных болезней обмена веществ, связанных с нарушением метаболизма гликозаминогликанов (ГАГ), приводящее к поражению органов и тканей. Обусловлены данные заболевания мутациями генов, контролирующих процесс внутрилизосомного гидролиза макромолекул.
Причиной МПС I типа является мутация в гене, кодирующем лизосомный фермент альфа-L-идуронидазу. Тип наследования: аутосомно-рецессивный. Ген IDUA, кодирующий альфа-L-идуронидазу локализован в хромосомной области 4p16.3. Из-за снижения активности фермента происходит накопление различных типов ГАГ и развивается соматическая манифестация в виде лицевого дисморфизма, гепатоспленомегалии, поражения сердца, дыхательной системы, изменений скелета, неврологической симптоматики, гематологические и офтальмологические изменения. Вариабельность МПС определяется типом накапливаемого субстрата при недостаточной деградации ГАГ: при МПС I типа происходит накопление гепарансульфата и дерматансульфата. Дефицит альфа-L-идуронидазы может привести к развитию различных фенотипов болезни, обусловливая отличия в тяжести симптоматики. Выделяют три клинических фенотипа:
- синдром Гурлер (мукополисахаридоз I H - тяжёлая форма)
- синдром Шейе (мукополисахаридоз I S - лёгкая форма)
- синдром Гурлер-Шейе (мукополисахаридоз I H/S - промежуточная форма)
Мукополисахаридоз I типа - это панэтническое заболевание, его частота составляет 1:100000 новорожденных. Приблизительно 50%-80% пациентов имеют тяжелую форму заболевания. МПС I H/S синдром Гурлер-Шейе встречается с популяционной частотой 1:100000-1:500000 новорожденных; МПС I S синдром Шейе - 1:500000 новорожденных. Однако, нужно учитывать, что существует определенная погрешность в оценке распространенности различных фенотипов заболевания, что может быть связано с более частым выявлением именно тяжелых форм МПС I.
Особенности кодирования заболевания или состояния (группы заболеваний или состояний) по Международной статической классификации болезней и проблем, связанных со здоровьем: Согласно МКБ10, заболевание относится к классу IV, болезням эндокринной системы, расстройству питания и нарушению обмена веществ, E76.0 - Мукополисахаридоз I типа.
В настоящее время МПС I рассматривается как заболевание с континуумом клинических фенотипов, различающихся по возрасту манифестации, тяжести клинических проявлений и скорости прогрессирования заболевания у пациентов. Таким образом, довольно условно выделяют тяжёлую форму заболевания (синдром Гурлер), с манифестацией на первом году жизни, с прогрессирующей кардио-респираторной недостаточностью и ярко выраженной неврологической симптоматикой; и мягкую форму (синдромы Гурлер-Шейе и Шейе), при которой симптомы появляются в возрасте 4-10 лет и болезнь медленно прогрессирует, при этом некоторые пациенты доживают до взрослого возраста. С учётом традиционно используемой классификации, ниже приведены характерные клинические признаки для МПС I различных форм.
Диагноз МПС тип I устанавливается на основании совокупности: анамнестических данных, клинических данных, результатов лабораторного исследования (биохимического и молекулярно-генетического анализа). Дифференциальная диагностика проводится с другими типами МПС, альфа-маннозидозом, поздними формами ганглиозидозов, муколипидозом, неинфекционными полиартритами, эпифизарными дисплазиями. Пациентам с установленным диагнозом МПС I проводится динамическое наблюдение.
При осмотре необходимо обратить внимание на основные клинические проявления МПС I: грубые черты лица; низкорослость; тугоподвижность суставов; помутнение роговицы; гепатомегалия; спленомегалия; пахово-мошоночные и пупочные грыжи (двусторонние); сердечные шумы.
Лечение МПС тип I включает как патогенетическое лечение назначение ФЗТ, так и проведение симптоматической терапии. Ведение пациентов с МПС тип I предполагает мультидисциплинарный подход с участием врача-педиатра, врача-невролога, врача-генетика, врача-офтальмолога, врача-кардиолога/врача-детского кардиолога, врача-пульмонолога, врача-сурдолога-оториноларинголога, врача-оториноларинголога, врача-хирурга/врача-детского хирурга, врача-челюстно-лицевого хирурга, врача-нейрохирурга, врача-физиотерапевта, медицинского психолога и врачей других специальностей, имеющих опыт в лечении этого редкого заболевания.
Специфической реабилитации пациентам с МПС I не требуется. В круг реабилитационных мероприятий пациентам с МПС I могут быть включены занятия с медицинским психологом, отдых в специализированных санаториях, а также социальная адаптация с участием специалистов и социальных работников, курсы массажа. Специфические методы реабилитации при наличии осложнений указаны в соответствующих разделах.
При проведении наркоза и интубации необходимо помнить о высоком риске компрессии спинного мозга вследствие нестабильности атлантоаксиального сустава. Короткая шея, ограничение подвижности нижней челюсти, увеличение языка, выраженная гипертрофия аденоидов и миндалин создают проблемы при проведении анестезиологического пособия, поэтому предпочтение следует отдавать местному или региональному обезболиванию. Пациент предварительно консультируется врачом-кардиологом/врачом детским кардиологом, врачом-оториноларингологом, врачом-анестезиологом-реаниматологом, врачом-неврологом. Обязательно проведение полного кардиологического обследования, полисомнографии (для выявления степени дыхательных нарушений), при необходимости - эндоскопии носоглотки и компьютерной томографии легких. Оперативное вмешательство с анестезией необходимо проводить в крупных медицинских центрах, имеющих отделение реанимации и интенсивной терапии (ОРИТ), так как интубация и последующая экстубация у таких пациентов может вызвать затруднения.
Синдром Гурлер или мукополисахаридоз I (МПС I) - одна из первых описанных форм мукополисахаридозов (МПС). Заболевание, изначально названное болезнь Пфаундлера-Гурлер, впервые описано двумя педиатрами: австрийским - Гертрудой Гурлер (1889-1965) и немецким - Пфаундлер Мейнард (1872-1947). Затем американским офтальмологом Шейе (1909-1990) описана вторая форма болезни с более поздним началом и более доброкачественным течением, названная синдром Шейе. Позже описана промежуточная форма болезни, названная синдром Гурлер-Шейе. МПС I - очень редкое заболевание. По оценкам специалистов, оно встречается всего лишь у одного из 100000 новорожденных. За прошедшие годы был создан специальный препарат, который позволяет замедлить прогрессирование болезни, смягчить некоторые из её проявлений. Однако, наряду с применением этого препарата, необходимо не забывать, о симптоматической терапии, физиотерапии, реабилитации, а главное - позитивном отношении к жизни пациента и членов его семьи.
Мукополисахаридоз I типа - относится к наследственным заболеваниям и наследуется по аутосомно-рецессивному типу. Это значит, что болезнь проявляется только в том случае, если оба родителя являются носителями болезни и, хотя сами они не болеют, передают ребёнку два пораженных гена. Большинство семей, где есть ребенок с этим заболеванием, не сталкивались раньше с подобной проблемой. Риск повторного рождения ребенка с МПС I в семье, где уже есть больные дети, составляет 25% на каждую беременность. Все семьи с МПС I должны обязательно пройти медико-генетическое консультирование и получить полную информацию от врача-генетика о риске повторного проявления данного заболевания в семье, обсудить со специалистом все вопросы, связанные с наследованием заболевания. В России медико-генетические консультации работают в каждом регионе.
Врачи на основании клинических симптомов могут заподозрить болезнь. Затем проводятся лабораторные тесты и инструментальное исследование. Поскольку разные типы МПС очень похожи по своим клиническим проявлениям, необходимо подтвердить диагноз с помощью лабораторных методов. Подтверждающая диагностика МПС заключается в определении уровня экскреции ГАГ в моче и измерении активности ферментов в клетках крови, пятнах высушенной крови или культуре кожных фибробластов. Для МПС I проводят определение активности альфа-L-идуронидазы. В дальнейшем рекомендуется проведение ДНК диагностики (если активность фермента была снижена).
Поставить диагноз синдрома Гурлер-Шейе новорожденному практически невозможно, так как наши пациенты рождаются в срок, с нормальными росто-весовыми показателями. Но при синдроме Гурлер - более тяжелой форме - первые клинические признаки заболевания появляются на первом году жизни. В ряде случаев, уже с рождения наблюдаются незначительное увеличение печени, пупочные или пахово-мошоночные грыжи. Рост замедляется, когда малыши достигают возраста одного-двух лет. Пациенты с тяжелой формой заболевания обычно почти прекращают расти в возрасте восьми лет - их рост обычно не превышает 110 см. Другие пациенты продолжают расти до подросткового возраста и достигают 152-160 см. При легкой форме рост у пациентов, как правило, ниже чем у здоровых сверстников, но бывает и почти нормальным.
Внешний вид пациентов с тяжёлой формой синдрома Гурлер необычен - они больше похожи друг на друга, чем на своих родителей и здоровых братьев и сестер. Изменения в строении их лиц обозначают специальным термином лицевой дизморфизм (огрубление черт): крупная голова, короткая шея, круглое лицо, широкий нос с широкой и плоской переносицей. При лёгкой и промежуточной форме заболевания внешние особенности у пациента столь незначительны, что их видят только врачи, а близкие и знакомые не замечают ничего необычного. У пациентов с МПС I из-за отложения мукополисахаридов кожа "толстая" и жёсткая, что затрудняет забор крови.
|